Welcome to the VizBin page.
You can find news and updates about our application for human-augmented binning here.
- 22/10/2015 - Awesome logo added. Thanks a lot to Linda Wampach (designer of the mothur logo)
- 04/02/2015 - Gallery added: In recent years, several metagenomic datasets have been created and published. To demonstrate the versatility, efficiency, and potential of VizBin beyond the original publication, we make visualizations (of at least a bigger subset) of these datasets available for the community in our Gallery. The addition of more "works" is also envisioned!
- 20/01/2015 - Manuscript published: Great news! The Software article manuscript VizBin - an application for reference-independent visualization and human-augmented binning of metagenomic data is published in Microbiome!!! Happy reading.
A description of VizBin, the underlying concept etc. can be found below.
Stay tuned for updates!
The application.
The "Download app" button always points to the most recent version of VizBin. The version of VizBin that is part of the manuscript can be downloaded here.
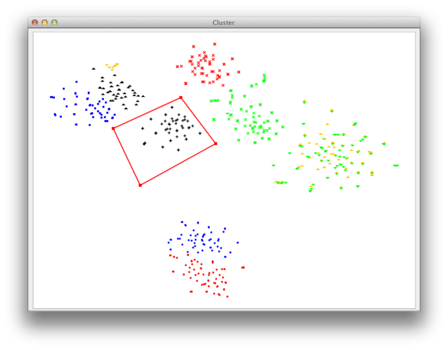
Below, you can see the screenshot of the visualization window. Each point represents a genomic fragment (here of length >= 1,000nt). The colors are added for visualization purposes only and each color represents an invdividual taxon of origin. The points contained in the red polygon are selected.
Usage.
VizBin is designed with the user in mind. Hence, we kept the default user-interface simple. No fiddling with parameters. All that is needed is a fasta file containing the sequences of interest. A step-by-step guide on using VizBin - including a description of loading the data, selecting points, and exporting the sequences represented by the selected points - is provided on the tutorial page of VizBin's github wiki.
We suggest to visualize fragments that are at least 1,000nt long. Visualizing shorter fragments is also possible but may lead to blurring of distinct clusters, hence the suggested threshold. Filtering sequences based on their length can be done directly from within VizBin, as we explain in the tutorial including filtering. Thus, no prior separate filtering step is required.
Example datasets
The concept.
We explain the concept underlying the intuitive visualization in detail in the publication Alignment-free Visualization of Metagenomic Data by Nonlinear Dimension Reduction. We also include experiments and results on datasets of varying complexity and size.
Gist
The gist of the concept is that VizBin uses the state-of-the-art nonlinear dimension reduction algorithm BH-SNE and appropriate data transformation to visualize (assembled) metagenomic data-inherent clusters.
Speed
Due to the efficiency of BH-SNE, VizBin can be used on regular desktop computers and returns results very fast (for 30,000 datapoints, it takes less than 4 minutes on a MacBook Pro from 2011). To achieve further speedups, we have integrated parallelization through the OpenMP API thus allowing to use multiple cores which are included in most modern computers.
Acknowledgments
Many people have collaborated on making this application become a reality: Cedric C. Laczny, Tomasz Sternal, Valentin Plugaru, Piotr Gawron, Arash Atashpendar, Hera Houry Margossian, Sergio Coronado, Laurens van der Maaten, Nikos Vlassis, and Paul Wilmes.
The present work was supported by an ATTRACT programme grant (A09/03) and a European Union Joint Programming in Neurodegenerative Diseases grant (INTER/JPND/12/01) to Paul Wilmes and an Aide à la Formation Recherche grant (AFR PHD/4964712) to Cedric C. Laczny all funded by the Luxembourg National Research Fund (FNR).

